¡Saludos a todos los exploradores del vasto océano de la salud renal! Hoy nos aventuramos en las profundidades del Síndrome de Alport, una condición que puede hacer que tus riñones sean el epicentro de una tormenta genética. Pero, ¿te has detenido a considerar las fascinantes historias que esta enfermedad tiene para contarnos? Prepárate para zarpar en un viaje de descubrimiento mientras desvelamos los misterios y curiosidades que rodean al Síndrome de Alport. 🌊💼
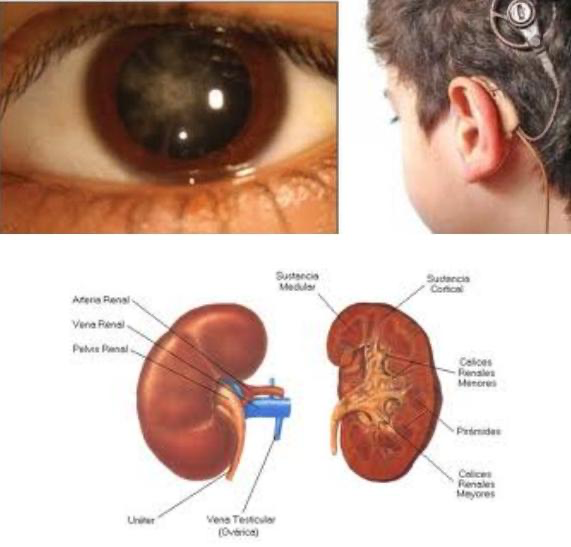
El síndrome de Alport es un trastorno hereditario poco frecuente que causa daño a los diminutos vasos sanguíneos en los riñones. También puede causar pérdida de la audición y problemas oculares.
El síndrome de Alport es una forma hereditaria de inflamación del riñón (nefritis). Es causado por un defecto (mutación) en un gen para una proteína en el tejido conectivo, llamada colágeno.
El trastorno poco común. Existen tres tipos genéticos:
Síndrome de Alport ligado al cromosoma X (XLAS, por sus siglas en inglés): Este es el tipo más común. La enfermedad es más grave en hombres que en mujeres.
Síndrome de Alport autosómico recesivo (ARAS, por sus siglas en inglés): La enfermedad es igual de grave en hombres y mujeres.
Síndrome de Alport autosómico dominante (ADAS, por sus siglas en inglés): Este es el tipo menos frecuente. La enfermedad es igual de grave en hombres y mujeres.
En todos los tipos de síndrome de Alport, los riñones resultan afectados. Los diminutos vasos sanguíneos en los glomérulos de los riñones resultan dañados. Los glomérulos filtran la sangre para producir orina y eliminar los productos de desecho de la sangre.
Al principio, no hay síntomas. Con el tiempo, a medida que los glomérulos resultan más y más dañados, se pierde la función renal y hay una acumulación de líquidos y productos de desecho en el cuerpo. La afección puede progresar hasta convertirse en enfermedad renal terminal (ERT) a una edad temprana, entre la adolescencia y los 40 años de edad. En ese punto, es necesario someterse a diálisis o a un trasplante de riñón.
Más información:


.jpg)

No hay comentarios:
Publicar un comentario